FlowSOM Clustering#
In this tutorial we are going to review how you can use FlowSOM to cluster single cell cytometry data. To demonstrate the FlowSOM algorithm we will be using the Levine 32 dimensional mass cytometry data originally introduced in the Phenograph paper “Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis” in 2015 (https://pubmed.ncbi.nlm.nih.gov/26095251/). You can obtain the Levine data from this GitHub repository: https://github.com/lmweber/benchmark-data-Levine-32-dim/tree/master/data.
import scanpy as sc
import anndata as ann
import numpy as np
import scipy as sp
import pandas as pd
import matplotlib.pyplot as pl
from matplotlib import rcParams
from matplotlib import colors
import seaborn as sb
import datetime
import pytometry as pm
sc.logging.print_versions()
sc.settings.verbosity = 3
-----
anndata 0.9.2
scanpy 1.9.5
-----
PIL 10.0.1
anyio NA
argcomplete NA
arrow 1.3.0
asttokens NA
attr 23.1.0
attrs 23.1.0
babel 2.13.0
backcall 0.2.0
certifi 2023.07.22
cffi 1.16.0
charset_normalizer 3.3.0
cloudpickle 2.2.1
comm 0.1.4
consensusclustering NA
cycler 0.12.0
cython_runtime NA
dask 2023.9.3
datashader 0.15.2
datashape 0.5.2
dateutil 2.8.2
debugpy 1.8.0
decorator 5.1.1
defusedxml 0.7.1
entrypoints 0.4
exceptiongroup 1.1.3
executing 2.0.0
fastjsonschema NA
fcsparser 0.2.7
fqdn NA
fsspec 2023.9.2
h5py 3.9.0
idna 3.4
ipykernel 6.25.2
ipython_genutils 0.2.0
ipywidgets 8.1.1
isoduration NA
jedi 0.19.1
jinja2 3.1.2
joblib 1.3.2
json5 NA
jsonpointer 2.4
jsonschema 4.19.1
jsonschema_specifications NA
jupyter_events 0.8.0
jupyter_server 2.8.0
jupyterlab_server 2.25.0
kiwisolver 1.4.5
kneed 0.8.5
lamin_utils 0.11.4
llvmlite 0.41.0
markupsafe 2.1.3
matplotlib 3.8.0
minisom NA
mpl_toolkits NA
multipledispatch 0.6.0
natsort 8.4.0
nbformat 5.9.2
numba 0.58.0
numpy 1.25.2
overrides NA
packaging 23.2
pandas 1.5.3
param 1.13.0
parso 0.8.3
patsy 0.5.3
pexpect 4.8.0
pickleshare 0.7.5
platformdirs 3.11.0
prometheus_client NA
prompt_toolkit 3.0.39
psutil 5.9.5
ptyprocess 0.7.0
pure_eval 0.2.2
pyct 0.5.0
pydev_ipython NA
pydevconsole NA
pydevd 2.9.5
pydevd_file_utils NA
pydevd_plugins NA
pydevd_tracing NA
pygments 2.16.1
pyparsing 3.1.1
pythonjsonlogger NA
pytometry 0.1.4
pytz 2023.3.post1
readfcs 1.1.7
referencing NA
requests 2.31.0
rfc3339_validator 0.1.4
rfc3986_validator 0.1.1
rpds NA
scipy 1.11.3
seaborn 0.12.2
send2trash NA
session_info 1.0.0
setuptools 67.6.0
sitecustomize NA
six 1.16.0
sklearn 1.3.1
sniffio 1.3.0
sphinxcontrib NA
stack_data 0.6.3
statsmodels 0.14.0
threadpoolctl 3.2.0
tlz 0.12.0
toolz 0.12.0
tornado 6.3.3
tqdm 4.66.1
traitlets 5.11.2
typing_extensions NA
uri_template NA
urllib3 2.0.6
wcwidth 0.2.8
webcolors 1.13
websocket 1.6.4
xarray 2023.9.0
yaml 6.0.1
zipp NA
zmq 24.0.1
zoneinfo NA
-----
IPython 8.16.1
jupyter_client 7.4.9
jupyter_core 5.3.2
jupyterlab 4.0.7
notebook 6.5.6
-----
Python 3.10.12 (main, Jun 11 2023, 05:26:28) [GCC 11.4.0]
Linux-6.2.0-36-generic-x86_64-with-glibc2.35
-----
Session information updated at 2023-11-07 08:47
First we will load the data.
adata = pm.io.read_fcs("./Levine_32dim_notransform.fcs")
adata.var
| n | channel | marker | $PnB | $PnE | $PnR | |
|---|---|---|---|---|---|---|
| Time | 1 | Time | 32 | 0,0 | 1024 | |
| Cell_length | 2 | Cell_length | 32 | 0,0 | 1024 | |
| DNA1 | 3 | DNA1 | 32 | 0,0 | 1024 | |
| DNA2 | 4 | DNA2 | 32 | 0,0 | 1024 | |
| CD45RA | 5 | CD45RA | 32 | 0,0 | 1024 | |
| CD133 | 6 | CD133 | 32 | 0,0 | 1024 | |
| CD19 | 7 | CD19 | 32 | 0,0 | 1024 | |
| CD22 | 8 | CD22 | 32 | 0,0 | 1024 | |
| CD11b | 9 | CD11b | 32 | 0,0 | 1024 | |
| CD4 | 10 | CD4 | 32 | 0,0 | 1024 | |
| CD8 | 11 | CD8 | 32 | 0,0 | 1024 | |
| CD34 | 12 | CD34 | 32 | 0,0 | 1024 | |
| Flt3 | 13 | Flt3 | 32 | 0,0 | 1024 | |
| CD20 | 14 | CD20 | 32 | 0,0 | 1024 | |
| CXCR4 | 15 | CXCR4 | 32 | 0,0 | 1024 | |
| CD235ab | 16 | CD235ab | 32 | 0,0 | 1024 | |
| CD45 | 17 | CD45 | 32 | 0,0 | 1024 | |
| CD123 | 18 | CD123 | 32 | 0,0 | 1024 | |
| CD321 | 19 | CD321 | 32 | 0,0 | 1024 | |
| CD14 | 20 | CD14 | 32 | 0,0 | 1024 | |
| CD33 | 21 | CD33 | 32 | 0,0 | 1024 | |
| CD47 | 22 | CD47 | 32 | 0,0 | 1024 | |
| CD11c | 23 | CD11c | 32 | 0,0 | 1024 | |
| CD7 | 24 | CD7 | 32 | 0,0 | 1024 | |
| CD15 | 25 | CD15 | 32 | 0,0 | 1024 | |
| CD16 | 26 | CD16 | 32 | 0,0 | 1024 | |
| CD44 | 27 | CD44 | 32 | 0,0 | 1024 | |
| CD38 | 28 | CD38 | 32 | 0,0 | 1024 | |
| CD13 | 29 | CD13 | 32 | 0,0 | 1024 | |
| CD3 | 30 | CD3 | 32 | 0,0 | 1024 | |
| CD61 | 31 | CD61 | 32 | 0,0 | 1024 | |
| CD117 | 32 | CD117 | 32 | 0,0 | 1024 | |
| CD49d | 33 | CD49d | 32 | 0,0 | 1024 | |
| HLA-DR | 34 | HLA-DR | 32 | 0,0 | 1024 | |
| CD64 | 35 | CD64 | 32 | 0,0 | 1024 | |
| CD41 | 36 | CD41 | 32 | 0,0 | 1024 | |
| Viability | 37 | Viability | 32 | 0,0 | 1024 | |
| file_number | 38 | file_number | 32 | 0,0 | 1024 | |
| event_number | 39 | event_number | 32 | 0,0 | 1024 | |
| label | 40 | label | 32 | 0,0 | 1024 | |
| individual | 41 | individual | 32 | 0,0 | 1024 |
As you can see, the dataset consists of numerous channels and some meta-variables such as individual and label.
adata[:, "label"].to_df()["label"].value_counts(dropna=False)
NaN 161443
7.0 26366
10.0 21099
8.0 20108
9.0 16520
13.0 6135
2.0 3905
4.0 3295
3.0 2248
11.0 1238
1.0 1207
6.0 916
14.0 513
12.0 330
5.0 304
Name: label, dtype: int64
adata[:, "individual"].to_df()["individual"].value_counts(dropna=False)
1.0 191351
2.0 74276
Name: individual, dtype: int64
channels = [
"DNA1",
"DNA2",
"CD45RA",
"CD133",
"CD19",
"CD22",
"CD11b",
"CD4",
"CD8",
"CD34",
"Flt3",
"CD20",
"CXCR4",
"CD235ab",
"CD45",
"CD123",
"CD321",
"CD14",
"CD33",
"CD47",
"CD11c",
"CD7",
"CD15",
"CD16",
"CD44",
"CD38",
"CD13",
"CD3",
"CD61",
"CD117",
"CD49d",
"HLA-DR",
"CD64",
"CD41",
]
We want to normalise the channels with a hyperbolic inverse sine with cofactor 5 (as performed in the original paper)
adata_arcsinh = pm.tl.normalize_arcsinh(adata[:, channels], cofactor=5, inplace=False)
We can now cluster our data with the flowsom_clustering function from the tl (tools) module of pytometry. The FlowSOM algorithm works in the following stages:
A self-organising map is trained that learns a low dimensional embedding of the single cell feature space
Consensus clustering (https://github.com/burtonrj/consensusclustering) of the SOM nodes identifies the optimal number of clusters to retain
Each observation is assigned to a cluster according to its nearest meta-cluster in the SOM
The function is going to take as input:
adata- our annotated dataframe of normalised channelskey_added- the key to add the cluster labels undersom_dim- the size of the self-organising map; (10, 10) is the default size used by the original FlowSOM implementation and usually does not need to be alteredsigma- this controls the radius of the neighbourhood function and again the default value of 1.0 works well in most caseslearning_rate- controls the size of weight vector in learning of SOM and the default value of 0.5 works well in most casesbatch_size- adjust if you have memory constraintsseed- random seed for reproducibilitymin_clusters- the minimum number of clusters when searching for the optimal number of clusters in consensus clusteringmax_clusters- the maximum number of clusters when searching for the optimal number of clusters in consensus clusteringreturn_clustering_objs- if True, the SOM and Consensus Clustering objects are returned; this can be helpful for inspecting the results as shown laterverbose- show progress bars etccopy- copy theadataobject (recommended)agglomerative_clustering_kwargs- keyword arguments passed to theAgglomerativeClusteringScikit-Learn object used by the consensus clustering
adata_flowsom, cluster_objs = pm.tl.flowsom_clustering(
adata=adata_arcsinh,
key_added="flowsom_clusters",
som_dim=(10, 10),
sigma=1.0,
learning_rate=0.5,
batch_size=500,
seed=42,
min_clusters=10,
max_clusters=20,
return_clustering_objs=True,
verbose=True,
inplace=True,
agglomerative_clustering_kwargs={"metric": "euclidean", "linkage": "ward"},
)
[ 500 / 500 ] 100% - 0:00:00 left
quantization error: 3.422194930315085
adata_flowsom.obs["flowsom_clusters"].value_counts()
cluster_6 50159
cluster_7 41397
cluster_2 34354
cluster_3 31562
cluster_0 23688
cluster_10 19259
cluster_14 17600
cluster_8 10731
cluster_9 10146
cluster_1 9226
cluster_4 6871
cluster_12 4179
cluster_11 3970
cluster_5 1406
cluster_13 1079
Name: flowsom_clusters, dtype: int64
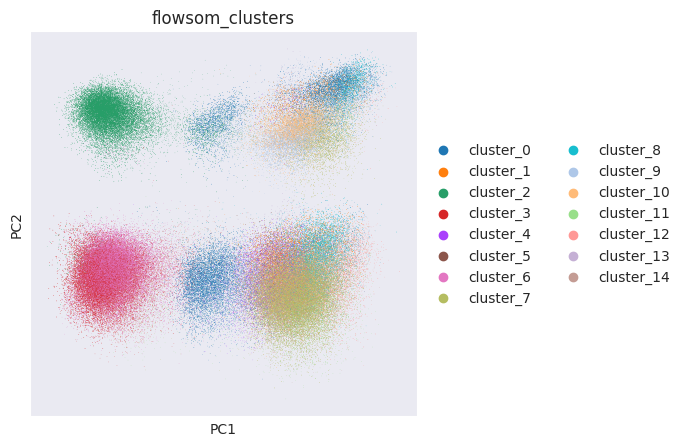
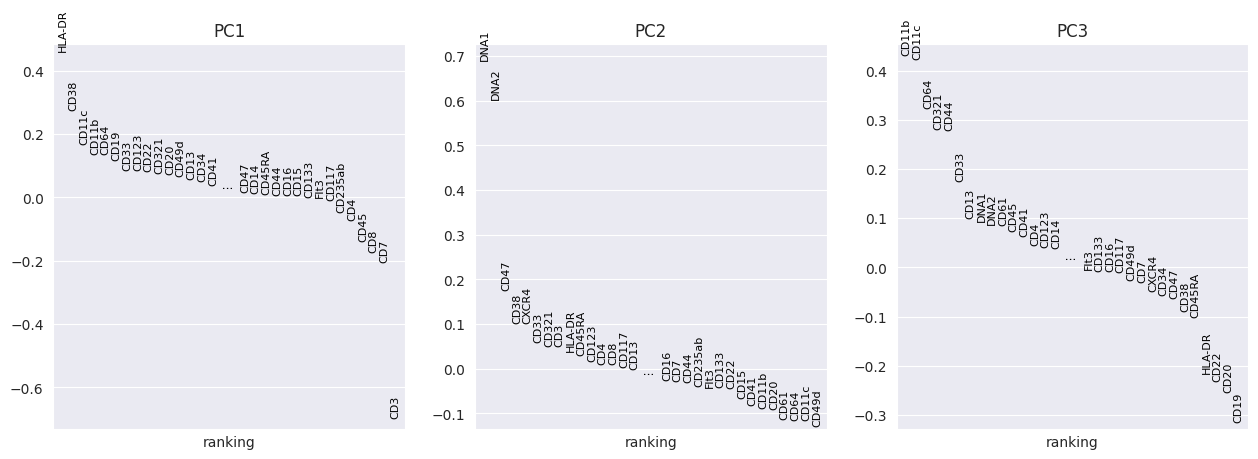
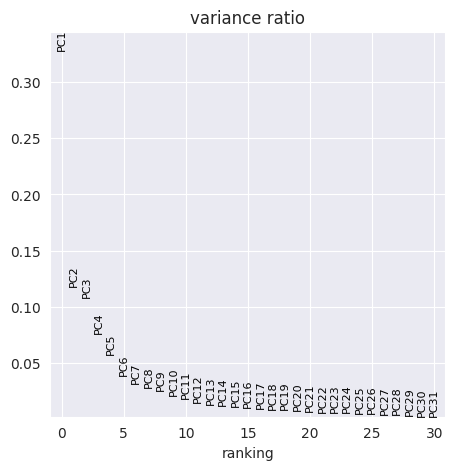
FlowSOM has generated 15 clusters. Lets visualise them using PCA and UMAP:
sc.pp.pca(adata_flowsom)
pl.rcParams["figure.figsize"] = (5, 5)
sc.pl.pca_overview(
adata_flowsom,
color="flowsom_clusters",
)
computing PCA
with n_comps=33
finished (0:00:00)
/home/ross/Projects/pytometry/venv/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:391: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
sc.pp.pca(adata_flowsom, n_comps=10)
sc.pp.neighbors(adata_flowsom, n_neighbors=15, use_rep="X_pca")
computing PCA
with n_comps=10
finished (0:00:00)
computing neighbors
/home/ross/Projects/pytometry/venv/lib/python3.10/site-packages/numba/np/ufunc/parallel.py:371: NumbaWarning: The TBB threading layer requires TBB version 2021 update 6 or later i.e., TBB_INTERFACE_VERSION >= 12060. Found TBB_INTERFACE_VERSION = 12050. The TBB threading layer is disabled.
warnings.warn(problem)
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:23)
sc.tl.umap(adata_flowsom)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:01:50)
sc.pl.umap(adata_flowsom, color="flowsom_clusters")
/home/ross/Projects/pytometry/venv/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:391: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(

We can inspect why 15 clusters were chosen by accessing the ConsensusClustering object returned by the our FlowSOM function. Consensus clustering works by calculating the consensus matrix of observations across random resamples of our data - stable clustering should result in observations consistently existing within the same or different clusters, if they sometimes appear in the same cluster or sometime do not, this suggests the clustering is unstable.
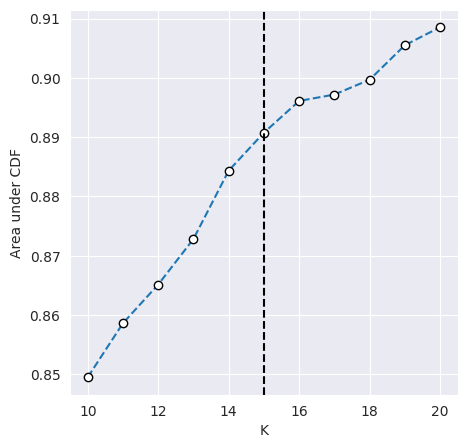
We can plot the CDF of the consensus matrix across different k clusters and observe the change point at which the area under the CDF no longer increased with the same magnitude. The plots below show the change point as detected by consensus clustering (first plot) and the CDF for each value of k (second plot).
cluster_objs["consensus_clustering"].plot_auc_cdf()
<Axes: xlabel='K', ylabel='Area under CDF'>
cluster_objs["consensus_clustering"].plot_cdf()
<Axes: xlabel='Consensus index value', ylabel='CDF'>
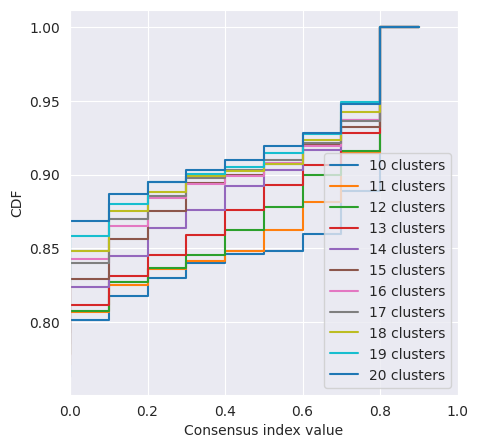
The change in CDF below and above a k of 15 is subtle and we would expect ideal clustering to have consensus index values occuring around 0 or 1, but biological data is often noisy and consensus clusters can be difficult to identify.
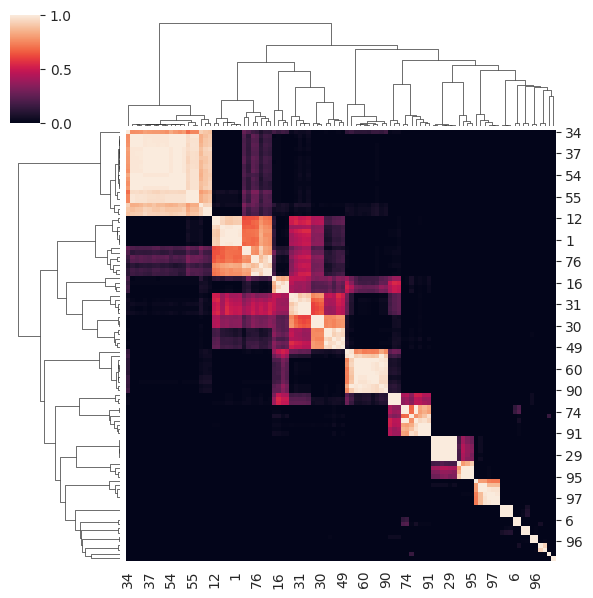
The consensus matrix for the chosen k of 15 is shown below. Notice how for many observations a clear consensus is established along the diagonal but some observations are more difficult and have a ‘fuzzy’ allocation across clusters.
cluster_objs["consensus_clustering"].plot_clustermap(15, figsize=(6, 6))
/home/ross/Projects/pytometry/venv/lib/python3.10/site-packages/seaborn/matrix.py:560: UserWarning: Clustering large matrix with scipy. Installing `fastcluster` may give better performance.
warnings.warn(msg)
/home/ross/Projects/pytometry/venv/lib/python3.10/site-packages/seaborn/matrix.py:560: UserWarning: Clustering large matrix with scipy. Installing `fastcluster` may give better performance.
warnings.warn(msg)
<seaborn.matrix.ClusterGrid at 0x7f50b5846740>